Chem 336
Spring 2006
Final Exam Questions
The following questions will be used for the final exam. On the day of the exam you will randomly select one of the questions and present your “best” solution for the analysis of the sample. In selecting the “best” you will want to consider both of the methods being considered. You should then be ready to outline as complete a method for analysis as possible, reflecting on linear/dynamic range, limits of quantitation, sample preparation, calibration methods and expected precision for both methods and why one would be preferred over the other.
1. East Gemini Lake at SJU can be called a “stew” of biological activity during the summer as a result of the increased nutrient levels present in the lakes from the waste water treatment plant. The nutrients that have been of greatest concern are phosphate and nitrate. The state of Minnesota as well as other states, many of major cities obtain their drinking water from surface water sources (rivers and lakes), as a result the EPA has set the maximum level for nitrate in drinking water at 10ppm.Previous analysis of East Gemini have shown levels of nitrate and phosphate from .0.5 – 50 ppm. You are faced with the choice of using classical spectrophotometric methods for phosphate (phosphomolybdate) and nitrate (cadmium reduction) or using capillary electrophoresis. Consider detection limits, linear dynamic range, calibration and sample preparation as you make your decision.
2. With each gathering for the Olympic games, competitors are required to submit both blood and urine samples for analysis for performance altering substances. Your company has contracted to do the analysis in Salt Lake City for the 2002 Winter Olympics. You are assigned to the analysis of steroidal drugs and their residues in these athletes. You will be doing random testing of about 10% of the athletes as well as blood and urine tests on all bronze, silver and gold metal winners. For this reason, the analysis must be relatively quick, but more importantly accurate. You have available to you both HPLC and GC-GCMS capabilities. Which method will you use. Consider specifically issues of sample matrix, internal standard vs external standard vs standard addition and separation.
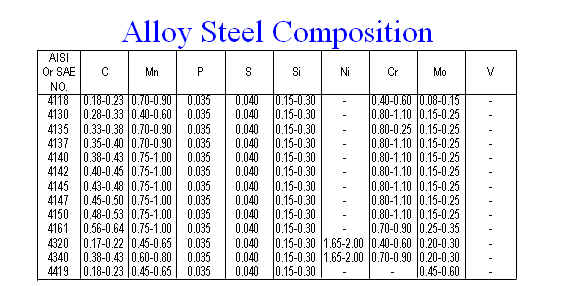
3. Though the analysis of steel is not glamorous, it is an analysis that must be done on many samples each day. The majority of these analyses are for trace elements that may be present in the steel, some that increase the strength and others that lead to premature corrosion and failure of the steel. You have been asked to develop a method that will allow for the analysis of chromium in steel. You have a flame atomic absorption spectrophotometer and a voltammeter as possible instruments to use. Given the general compositional make up of steel from the table below, (of course Fe makes up the remaining %) select the “best” method paying particular attention to interferences, detection limits and calibration methods.
numbers expressed in weight %
4. Hard water creates a number of difficulties in manufacturing, heating and even in drinking water systems. You need to develop a procedure for measuring water hardness (+2, +3 ions, specifically Ca, Mg, Fe) before and after processing for the removal of these ions.The source water can have ion concentrations as high as 180 ppm of CaCO3 while the “soft” water can be as low as 15 ppm. Your supervisor has just found the following water processing system and wants you to assess their claims. You decide you can use an ion selective electrode (ISE) or AA for the assessment. Considering the dynamic range, sample prep and detection limits specifically, select the “best” method.
 MAGNETICSOLUTIONSTM
MAGNETICSOLUTIONSTM
![]() Magnetic water treatment is simply the attachment of strong permanent magnets or
electromagnets to the incoming water pipes of your home. One or more
magnetic devices are clamped onto or into the water pipe leading from the water
meter or well to your house. The magnets surround the water pipe creating a strong
magnetic field within the pipe. The magnetic field interacts with positive and negative
ions associated with dissolved hard water minerals and other dissolved solids within the
water flowing through the pipe. The result is that the water complexes housing the
dissolved solids are disturbed, freeing the minerals and allowing them to serve as inflow
nucleation points for precipitating calcium. Since the calcium crystals nucleate on the
inflow particles and not the plumbing, scale is prevented or significantly reduced.
In addition, the water's surface tension and pH are reduced allowing the water to once
again develop a hunger for the buffering agent calcium carbonate. As a result, the water
flowing past plumbing with available calcium will allow previously adsorbed minerals
to reenter the liquid phase.
Magnetic water treatment is simply the attachment of strong permanent magnets or
electromagnets to the incoming water pipes of your home. One or more
magnetic devices are clamped onto or into the water pipe leading from the water
meter or well to your house. The magnets surround the water pipe creating a strong
magnetic field within the pipe. The magnetic field interacts with positive and negative
ions associated with dissolved hard water minerals and other dissolved solids within the
water flowing through the pipe. The result is that the water complexes housing the
dissolved solids are disturbed, freeing the minerals and allowing them to serve as inflow
nucleation points for precipitating calcium. Since the calcium crystals nucleate on the
inflow particles and not the plumbing, scale is prevented or significantly reduced.
In addition, the water's surface tension and pH are reduced allowing the water to once
again develop a hunger for the buffering agent calcium carbonate. As a result, the water
flowing past plumbing with available calcium will allow previously adsorbed minerals
to reenter the liquid phase.
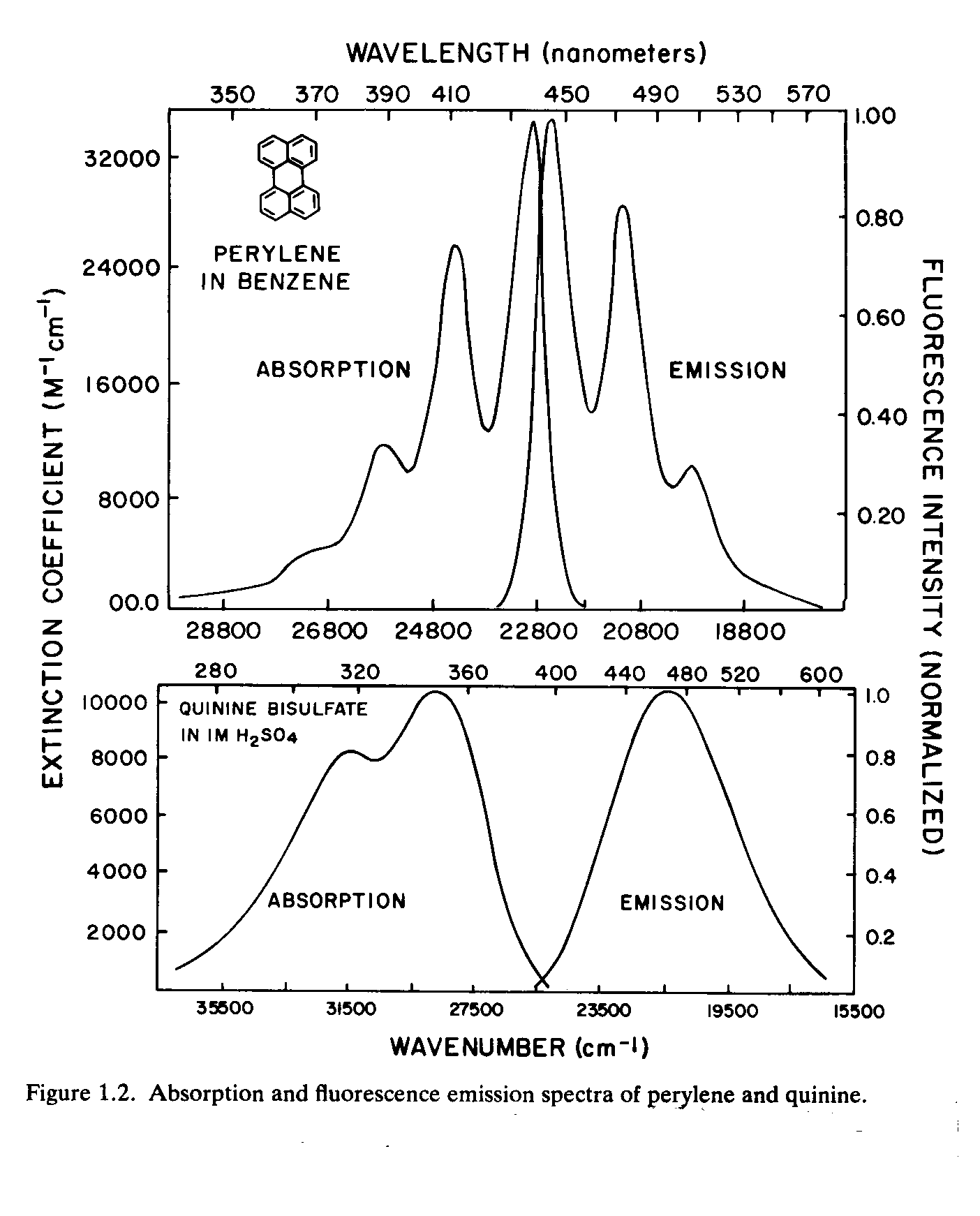
5. A manufacturer of quinine for tonic water fears that his feed stock might be contaminated by perylene, probably at the 1% or lower level. He has asked you to analyze this stock for the perylene and give a best estimate of the level. You have decided to use either HPLC with fluorescence detection or straight fluorescence using simultaneous spectrophotometric methods. (See spectra below). Concern yourself with both spectrophotometric and chromatographic resolution, interferences and separation.
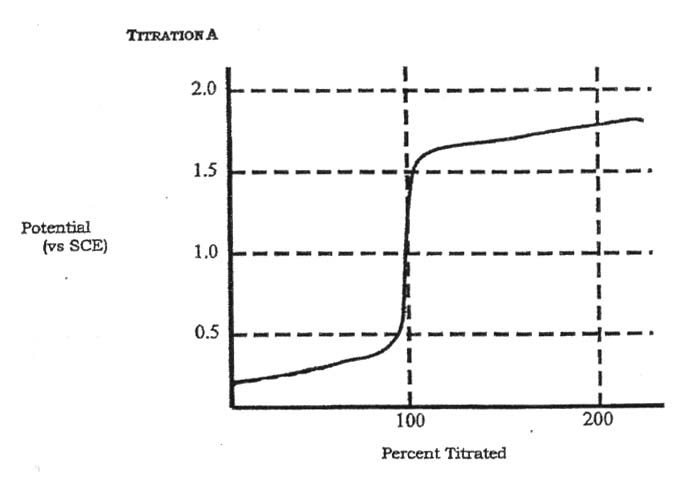
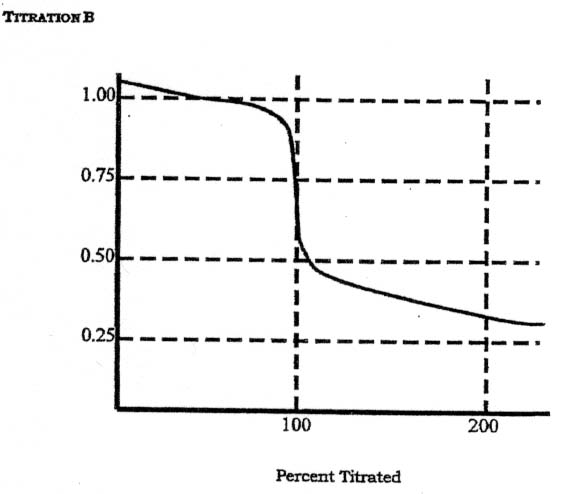
6. Below are two potentiometric titration curves obtained using a Pt indicator electrode and an SCE reference electrode. The half-reactions and formal electrode potentials for several redox couples are listed below:
|
half-reaction |
Eo' (vs SHE) |
|
H2O2 + 2H+ + 2e– = 2H2O
Ce+4 + e– = Ce+3
V(OH)4+ + 2H+ + e– = VO+2 + 3H2O
Fe+3 + e– = Fe+2
VO+2 + 2H+ + e– = V+3 + H2O
Sn+4 + 2e– = Sn+2 |
1.77
1.44
1.00
0.77
0.36
0.25 |
Assume that for each titration, the hydrogen ion concentration is buffered at 1 M and the sample and titrant solutions are also 1 M. For each curve, and using only the Table above,
a) Indicate which species (not half reaction) is: i) the titrant, ii) the sample
b) Write down the balanced chemical reaction for each titration.
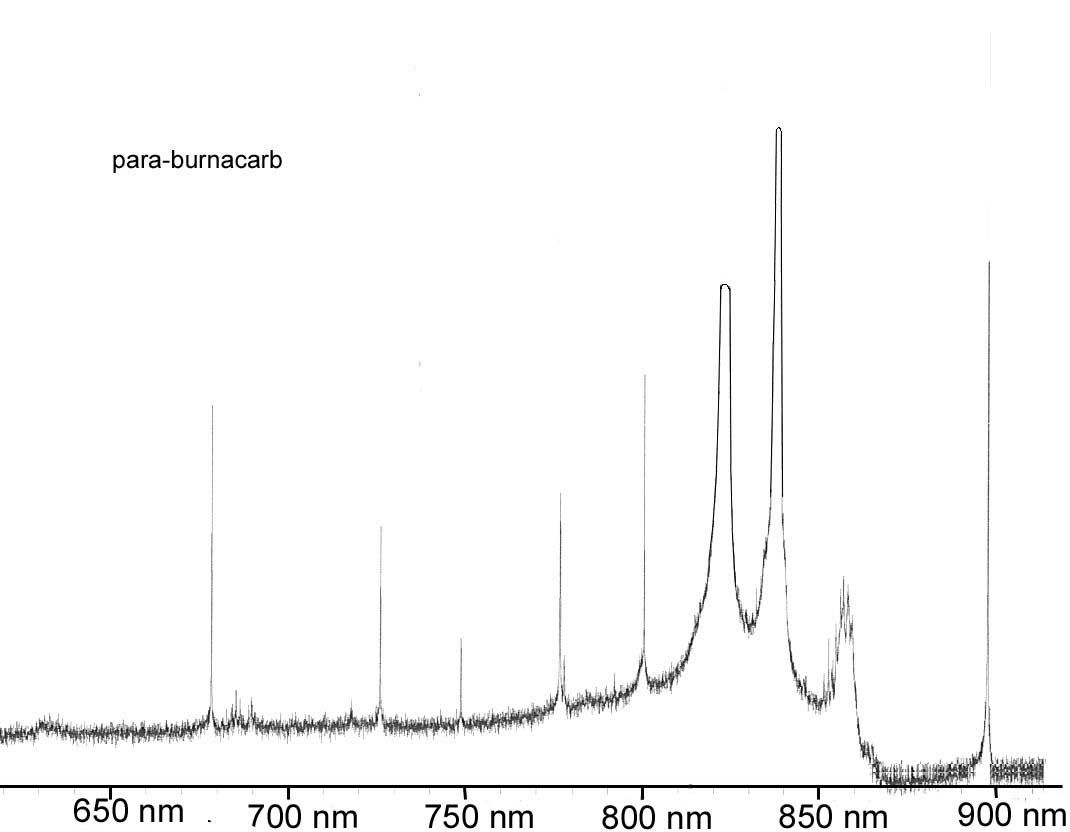
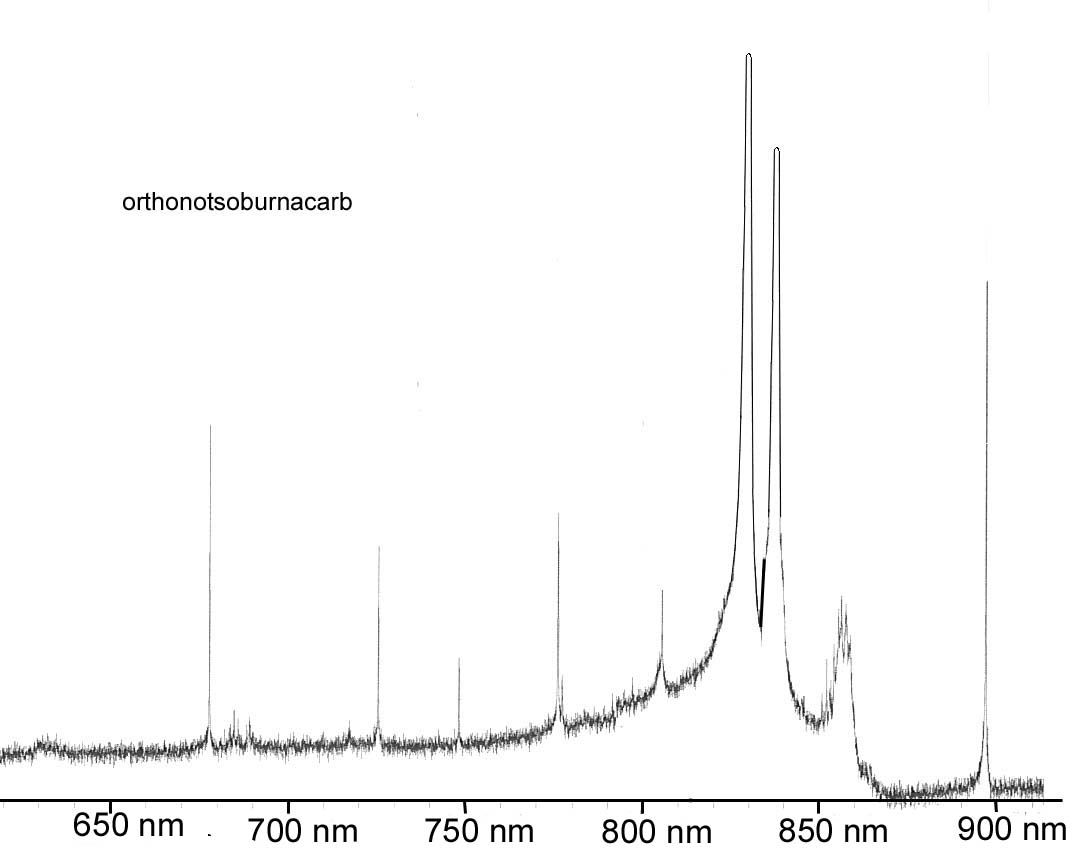
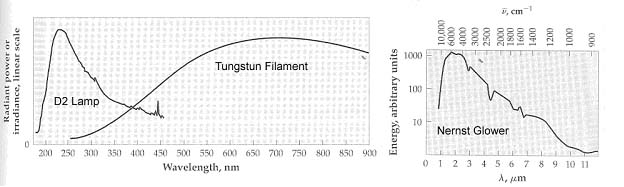
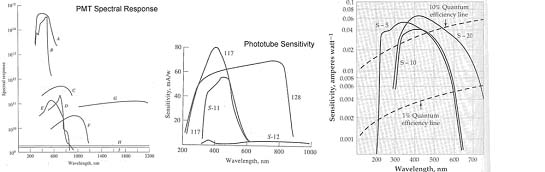
7. You have been approached by a colleague from the un-natural sciences to analyze a health food additive; para-burnacarb. The spectra for the compound is shown below. Unfortunately, the process of manufacturing the para-burnacarb creates an impurity of orthonotsoburnacarb with the spectra shown below as well. You recognize immediately that the HP Diode array UV-Vis is not going to cut it for the analysis (why) and decide to put together a simple system specific for this analysis. Given the information on sources and detectors below, select the appropriate radiation source and detector, establish the appropriate grating parameters and slit width to use, and the appropriate material for the sample cell for analyzing this product in a solution of your choice. We want to have an analysis which will give good sensitivity and selectivity for the para-burnacarb, and minimize interference by the orthonotsoburnacarb.
Sources
Detectors
8. The E½ for the reversible Sn+2, Sn+4 redox couple is –0.25 (vs SCE) in 1 M HCl on a platinum indicator electrode. Assuming the diffusion coefficients for both Sn+2 and Sn+4 are equal and that the residual currents are negligible, sketch the approximate current – voltage curves you would obtain using a SCE reference electrode and a platinum indicator electrode for (a) and (b) below. Be sure to label the voltage axis carefully. (NOTE: Assume the cathodic and anodic limits are – 0.45 volts and + 0.65 volts.)
a) a solution that is 1 M HCl and 1 x 10–4 M in Sn+2.
b) a solution that is 1 M HCl, 1 x 10–4 M in Sn+4 and 3 x 10–4 M in Sn+2.
9.
5. Consider the flame photometric determination of a metal which exhibits two emission lines of about equal intensity. (See table below.)
|
λ (nm) |
energy change (rE ) |
where: E0 = ground state E1 = 1st excited state E2 = 2nd excited state
|
|
500 |
E2 – E0 |
|
|
650 |
E2 – E1 |
Calibration curves for the two wavelengths were obtained using the same set of standard solutions. These are shown below:
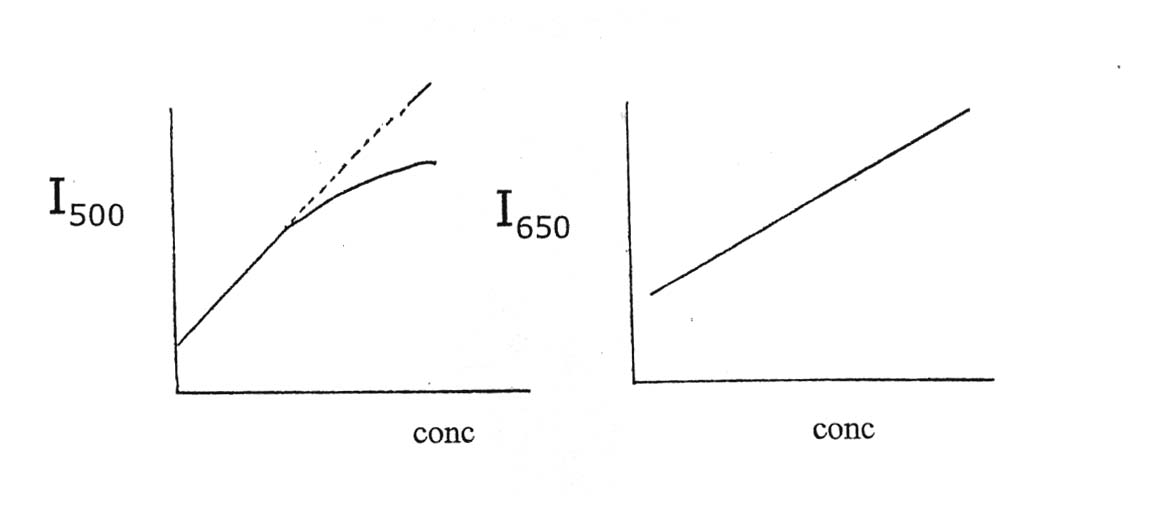
Explain, why the curve at 500 nm shows a large negative deviation and that at 650 nm does not. (HINT: What process causes the curve to bend over at high concentrations in flame photometry?)
10. Given the series of half-reactions and corresponding standard electrode potentials below,
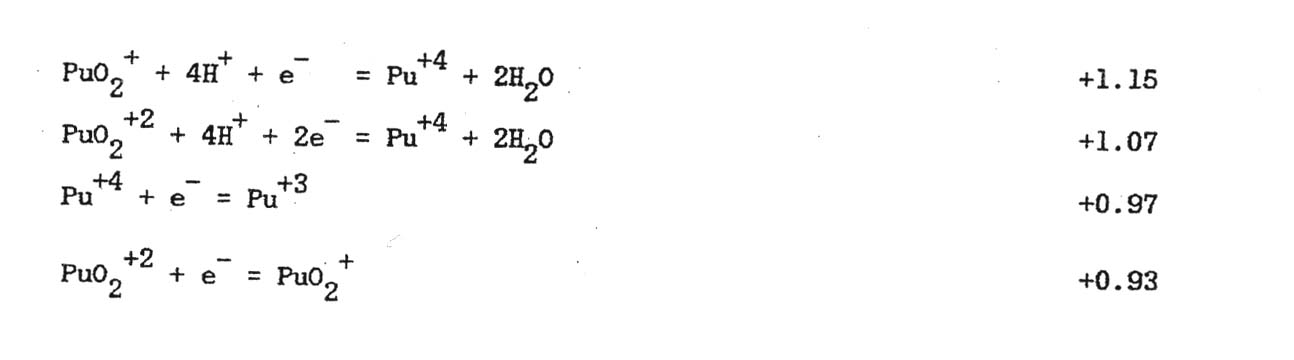
a. Explain how you might attempt to experimentally measure the standard electrode potential for the half reaction
PuO2+ + 4 H+ + 2 e- ↔ Pu3+ + 2 H2O
b. Explain why that would not work.
c. Show how you could calculate it.
11. At ordinary temperatures the acid ionization constant for the indicator HIn has a value of 5.40 x 10-7. Absorbance data (1.00-cm cells) for 5.00 x 10-4 M solutions of the indicator in strongly acidic and strongly alkaline media are given below.
Absorbance Absorbance
8 (nm) pH 1.00 pH 13.00 8 (nm) pH 1.00 pH 13.00
440 0.401 0.067 570 0.303 0.515
470 0.447 0.050 585 0.263 0.648
480 0.453 0.050 600 0.226 0.764
485 0.454 0.052 615 0.195 0.816
490 0.452 0.054 625 0.176 0.823
505 0.443 0.073 635 0.160 0.816
535 0.390 0.170 650 0.137 0.763
555 0.342 0.342 680 0.097 0.588
a) Predict the color of the acid form of the indicator HIn. Explain your reasoning.
b) What wavelength would be suitable for the spectrophotometric analysis of the indicator in its acidic form? Explain your reasoning.
c) What would be the absorbance of a 1.00 x 10-4 M solution of the indicator in its alkaline form when measured at 585 nm in a 2.00-cm cell? Show your work.
d) At what wavelength would the absorbance of the indicator be independent of pH? Explain your reasoning.
12. There are three major Atomic Spectroscopy Techniques: Atomic Absorption, Atomic Emission (either flame or ICP) and Atomic Fluorescence. Answer the following questions about these techniques:
a) What is the role of the hollow cathode lamp and “chopper” in atomic absorption spectrophotometry?
b) What is the importance of bandwidth in these optical methods.
c) What are the major differences between Atomic Emission and Atomic Fluorescence?
d) What are the major differences between conventional flame emission and ICP emission?
13. Two peaks emerge from a chromatography column as sketched in the illustration.
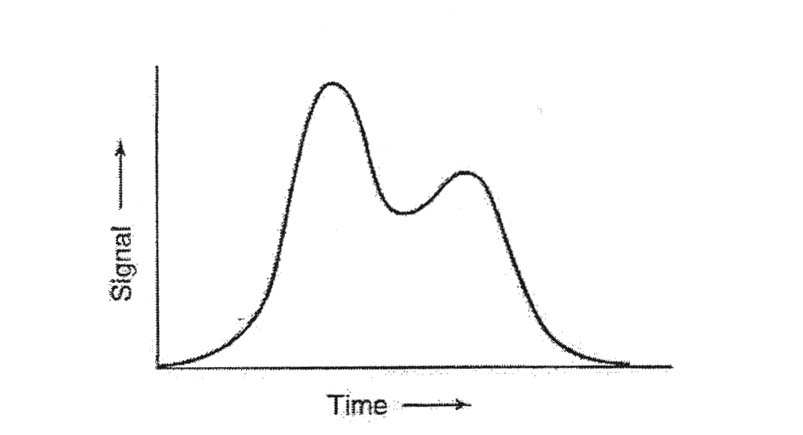
The resolution is given by resolution =
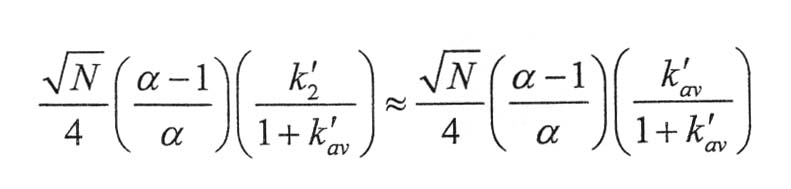
a) You can decrease the capacity factors by increasing the solvent strength. Sketch what the chromatogram would look like if N and α were kept the same, but k'av were increased. Sketch the chromatogram if k'av were decreased.
b) If you change the solvent or the stationary phase, you will change the relative retention, α. Sketch the chromatogram if N and k'av were kept the same but α were increased.
c) If you increase the column length or decrease the flow rate or particle size, you can increase the number of plates, N. Sketch the chromatogram if α and k'av were kept the same but N were increased.
14. Consider the extraction of from aqueous solution into organic solution by reaction with protonated ligand, HL:
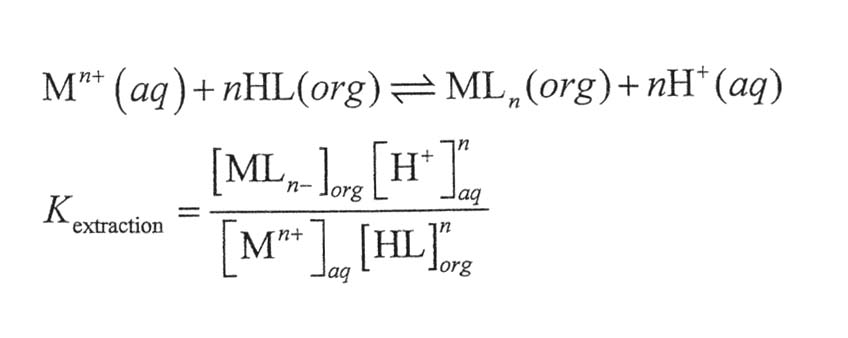
Rewrite the equation for D in terms of Kextraction and express Kextraction in terms of the constants. give a physical reason why each constant increases or decreases Kextraction.
15. a) When a solution containing 234 mg of butanol (MW 74.12) and 312 mg of hexanol (MW 102.17) in 10.0 mL was separated by gas chromatography, the relative peak areas were butanol:hexanol = 1.00:1.45. Considering butanol to be the internal standard, find the response factor for hexanol.
b) Use Equation 24-2 to estimate the areas of the peaks for butanol and hexanol in the gas chromatogram shown.
c) The solution from which the chromatogram was generated contained 112 mg of butanol. What mass of hexanol was in the solution?
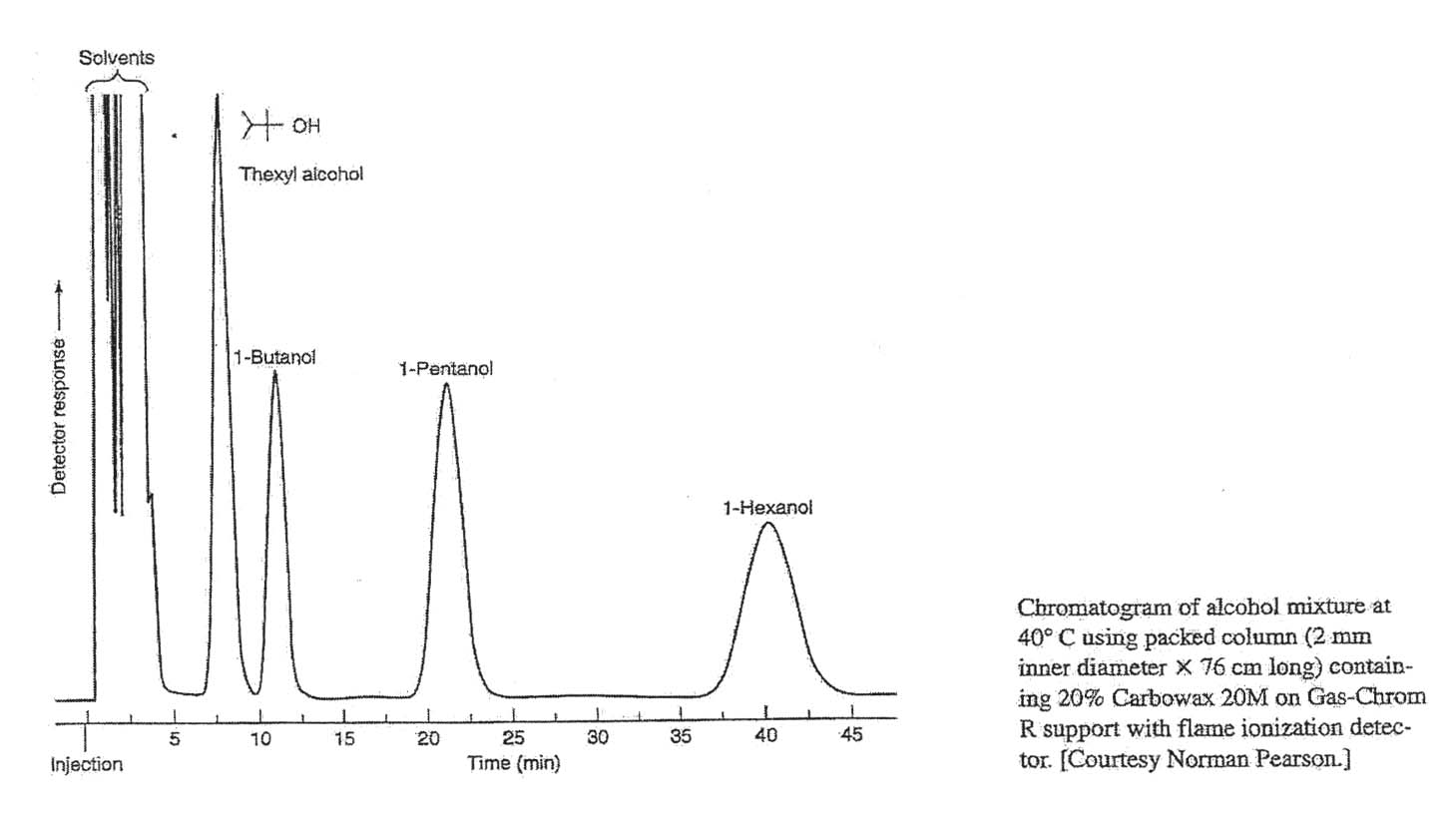
d) What is the largest source of uncertainty in this problem? How great is this uncertainty?
16. a) When you try separating an unknown mixture by reversed-phase chromatography with 50% acetonitrile/50% water, the peaks are too close together and are eluted in the range . Should you use a higher or lower concentration of acetonitrile in the next run?
b) When you try separating an unknown mixture by normal-phase chromatography with pure 50% hexane/50% methyl t-butyl ether, the peaks are too close together and are eluted in the range . Should you use a higher or lower concentration of hexane in the next run?