Biochemistry Online: An Approach Based on Chemical Logic
CHAPTER 7 - CATALYSIS
B: MECHANISMS OF ENZYME CATALYSIS
BIOCHEMISTRY - DR. JAKUBOWSKI
004/12/16
|
Learning Goals/Objectives for Chapter 7B: After class and this reading, students will be able to
|
We can apply what we learned about catalysis by small molecules to enzyme-catalyzed reactions. To understand the mechanism of an enzyme-catalyzed reaction, we try to alter as many variables, one at a time, and ascertain the effects of the changes on the activity of the enzyme. Kinetic methods can be used to obtain data from which inferences about the mechanism can be made. Obviously, crystal structures of the enzyme in the presence and absence of a competitive inhibitor give abundant information about possible mechanisms. It is amazing, however, how much information about enzyme mechanism can be gained even if all you have is a blender, a stopwatch, an impure enzyme, and a few substrates and inhibiting reagents. Systematically, the kineticist, medicinal chemist and molecular biologists (i.e. a well trained chemist) can change:
- the substrate - for example, changing the leaving group or acyl sustituents of a hydrolyzable substrate;
- the pH or ionic strength - which can give data about general acids/bases in the active site;
- the enzyme - by chemical modification of specific amino acids, or through site-specific mutagenesis;
- the solvent - as will be discussed in the next chapter section .
We will explore in detail the mechanisms of three enzymes. For carboxypeptidase, we will study possible mechanisms for the cleavage of C-terminal hydrophobic amino acids from a peptide. For lysozyme, we will study the structural features of the enzyme and substrates along with the mechanism for cleavage of glycosidic links in bacterial peptidoglycan cell walls. For chymotrypsin, we will study experiments which vary the substrate, pH, and the enzyme and infer from this information about a mechanism consistent with the experimental data. Kinetic analyses can be used to determine the:
- order of binding/dissociation of substrates and products
- rate constants for individual steps
- and clues to the nature of catalytic groups found in the enzyme.
B1. Carboxypeptidase
A peptide substrate binds at the active site of the enzyme. X-ray structures of the enzyme with and without a competitive inhibitor show a large conformational change at the active site when inhibitor or substrate is bound. Without inhibitor, several waters occupy the active site. When inhibitor and presumably substrate are bound, the water leaves (which is entropically favored), and Tyr 248 swings around from near the surface of the protein in the absence of a molecule in the active site to interact with the carboxyl group of the bound molecule, a distance of motion equal to about 1/4 the diameter of the protein. This effectively closes off the active site and expels the water. A Zn2+ ion is present at the active site. It is bound by His 69, His 196, Glu 72, and finally a water molecule as the fourth ligand. A hydrophobic pocket which interacts with the phenolic group of the substrate accounts for the specificity of the protein.
In the catalytic mechanism, Zn2+ helps polarize the labile amide bond, while Glu 270, acting as a general base, which along with Zn2+ helps promote dissociation of a proton from the bound water, making it a better nucleophile. Water attacks the electrophilic carbon of the sessile bond, with Glu 270 acting as a general base catalyst. The tetrahedral intermediate then collapses, expelling the leaving amine group, which picks up a proton from Glu 270, which now acts as a general acid catalyst. People used to believe that Tyr 248 acted as a general acid, but mutagenesis showed that Tyr 248 can be replaced with Phe 248 without significant effect on the rate of the reaction.
![]() Jmol:
Updated Carboxypeptidase A
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Carboxypeptidase A
Jmol14 (Java) |
JSMol (HTML5)
B2. Lysozyme
This enzyme, found in cells and secretions of vertebrates but also in viruses which infect bacteria, cleaves peptidoglycan GlcNAc (β 1->4) MurNAc repeat linkages (NAG-NAM) in the cell walls of bacteria and the GlcNAc (β 1->4) GlcNAc (poly-NAG) in chitin, found in the cells walls of certain fungi. Since these polymers are hydrophilic, the active site of the enzyme would be expected to contain a solvent-accessible channel into which the polymer could bind. The crystal structures of lysozyme and complexes of lysozyme and NAG have been solved to high resolution. The inhibitors and substrates form strong H bonds and some hydrophobic interactions with the enzyme cleft. Kinetic studies using (NAG)n polymers show a sharp increase in kcat as n increases from 4 to 5. The kcat for NAG6 and (NAG-NAM)3 are similar. Models studies have shown that for catalysis to occur, (NAG-NAM)3 binds to the active site with each sugar in the chair conformation except the fourth which is distorted to a half chair form, which labilizes the glycosidic link between the 4th and 5th sugars. Additional studies show that if the sugars that fit into the binding site are labeled A-F, then because of the bulky lactyl substituent on the NAM, residues C and E can not be NAM, which suggests that B, D and F must be NAM residues. Cleavage occurs between residues D and E.
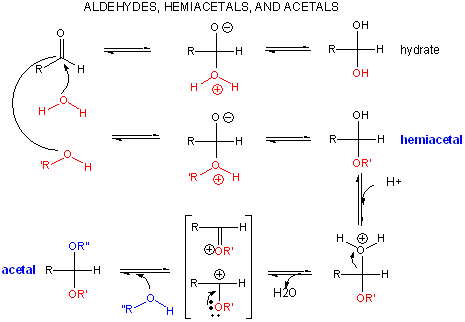
A review of the chemistry of glycosidic bond (an acetal) formation and cleavage shows the acetal cleavage is catalyzed by acids and proceeds by way of an oxonium ion which exists in resonance form as a carbocation.
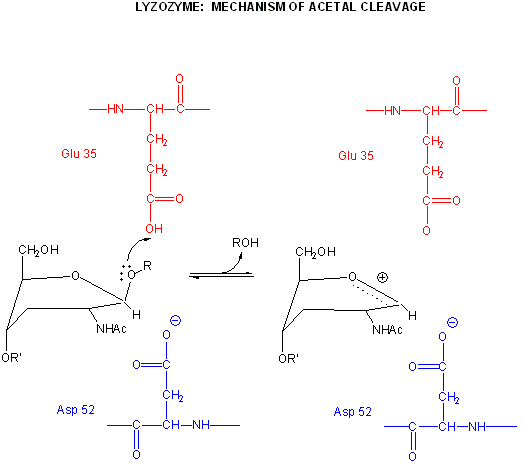
Catalysis by the enzyme involves Glu 35 and Asp 52 which are in the active site. Asp 52 is surrounded by polar groups but Glu 35 is in a hydrophobic environment. This should increase the apparent pKa of Glu 35, making it less likely to donate a proton and acquire a negative charge at low pH values, making it a better general acid at higher pH values. The general mechanism appears to involve:
-
binding of a hexasaccharide unit of the peptidoglycan with concomitant distortion of the D NAM.
-
protonation of the sessile acetal O by the general acid Glu 35 (with the elevated pKa), which facilitates cleavage of the glycosidic link and formation of the resonant stabilized oxonium ion.
-
Asp 52 stabilizes the positive oxonium through electrostatic catalysis. The distorted half-chair form of the D NAM stabilizes the oxonium which requires co-planarity of the substituents attached to the sp2 hybridized carbon of the carbocation resonant form (much like we saw with the planar peptide bond).
-
water attacks the stablized carbocation, forming the hemiacetal with release of the extra proton from water to the deprotonated Glu 35 reforming the general acid catalysis.
Binding and distortion of the D substituent of the substrate (to the half chair form as shown above) occurs before catalysis. Since this distortion helps stabilize the oxonium ion intermediate, it presumably stabilizes the transition state as well. Hence this enzyme appears to bind the transition state more tightly than the free, undistorted substrate, which is yet another method of catalysis.
pH studies show that side chains with pKa's of 3.5 and 6.3 are required for activity. These presumably correspond to Asp 52 and Glu 35, respectively. If the carboxy groups of lysozyme are chemically modified in the presence of a competitive inhibitor of the enzyme, the only protected carboxy groups are Asp 52 and Glu 35.
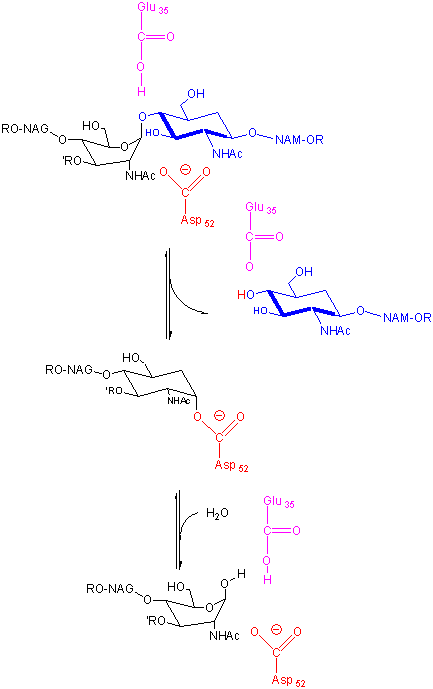
In an alternative mechanism, Asp 52 acts as a nucleophilic catalysis and forms a covalent bond with NAM, expelling a NAG leaving group with Glu 35 acting as a general acid. This alternative mechanism also is consistent with other β-glycosidic bond cleavage enzyme. Substrate distortion is also important in this alternative mechanism.
Figure: alternative mechanism
B3. Chymotrypsin
Chymotrypsin, a protease, cleaves amides as well as small ester substrates after aromatic residues. The following data using different chymotrypsin substrates suggests a covalent intermediate occurs on chymotrypsin catalyzed cleavage of esters and amides.
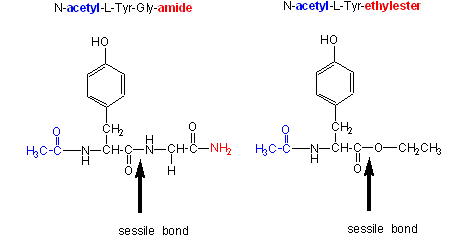
- changing the substrate - for example changing the leaving group or acyl sustituents of a hydrolyzable substrate:
| Chymotrypsin substrate cleavage, 25oC, pH 7.9 | |||
| kinetic constants | Acetyl-Tyr-Gly-amide | Acetyl-Tyr-O Ethylester | Ester/Amide |
| kcat (s-1) | 0.50 | 193 | 390 |
| Km (M) | 0.023 | 0.0007 | 0.03 |
| kcat/Km (M-1s-1) | 22 | 280,000 | 12,700 |
|
Kinetic constants for chymotrypsin cleavage of N-acetyl-L-Trp Derivatives - N-acetyl-L-Trp-X |
||
| X | kcat (s-1) | Km x 103 (M) |
| -OCH2CH3 | 27 | 0.097 |
| -OCH3 | 28 | 0.095 |
| -p-nitrophenol | 31 | 0.002 |
| -NH2 | 0.026 | 7.3 |
- the kcat and kcat/Km are larger and the Km smaller for ester substrates compared to amide substrates, suggesting that amides are more difficult to hydrolyze (tables 1 and 2 above). This is expected given the poorer leaving group of the amide.
- the kcat for the hydrolysis of ester substrates doesn't depend on the nature of the leaving group (i.e. whether it is a poorer leaving group such as methoxy or a better leaving group such as p-nitrophenolate) suggesting that this step is not the rate limiting step for ester cleavage. (Table 2). Without the enzyme, p-nitrophenyl esters are cleaved much more rapidly than methyl esters. Therefore deacylation must be rate limiting. But deacylation of what? If water was the nucleophile, release of the leaving group would result in both products, the free carboxyl group and the amine being formed simultaneously. This suggests an acyl-enzyme covalent intermediate.
- When the acyl end of the ester substrate is changed, without changing the leaving group (a p-nitrophenyl group), a covalent intermediate can be trapped. Specifically, the deacylation of a trimethyacetyl group is much slower than an acetyl group. It is so slow that a 14C-labeled trimethylacetyl-labeled chymotrypsin intermediate can be isolated after incubation of chymotrypsin with 14C-labeled p-nitrophenyltrimethylacetate using gel filtration chromatography.
We have seen a kinetic mechanism consistent with these ideas before. The reaction equations are shown below:
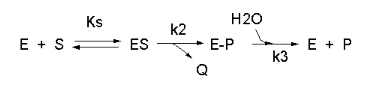
In this reaction, a substrate S might interact with E to form a complex, which then is cleaved to products P and Q. Q is released from the enzyme, but P might stay covalently attached, until it is expelled. This conforms exactly to the mechanism described above. For chymotrypsin-catalyzed cleavage, the step characterized by k2 is the acylation step (with release of the leaving group such as p-nitrophenol in Lab 5). The step characterized by k3 is the deacylation step in which water attacks the acyl enzyme to release product P (free phosphate in Lab 5). In class and for homework you derived the following equation::
Equation 7: v = [(k2k3)/(k2 + k3)]EoS/[Ks(k3)/(k2+k3)] + S
As mentioned above, for hydrolysis of ester substrates, which have better leaving groups compared to amides, deacylation is rate limiting, ( k3<<k2). Then equation 7 becomes
v = k3EoS/[Ks(k3)/(k2) + S]
Vm = k3Eo and Km = Ks(k3)/(k2)
For amide hydrolysis, as mentioned above, acylation can be rate-limiting (k2<<k3). Then equation 7 becomes:
v = k2EoS/[Ks+ S]
Vm = k2Eo and Km = Ks
Just as we saw before for the rapid equilibrium assumption (when ES falls apart to E + S more quickly than it goes to product), Km = Ks in amide hydrolysis.
- changing the pH or ionic strength - which can give data about general acids/bases in the active site:
- a graph of kcat as a function of pH indicates that a group of pKa of approx. 6 must be deprotonated to express activity (i.e. Vm/2 is at about pH 6). This suggests that an active site histidine is necessary, which if it must be deprotonated to express activity, must be acting as a general base.
- a graph of kcat/Km shows a bell-shaped curve indicating the necessity of a deprotonated side chain with a pKa of about 6 (i.e. the same His above) and a group which must be protonated with a pKa of about 10. This turns out to be an N terminal Ile (actually at the 16 position in the inactive precursor of chymotrypsin called chymotrypsinogen, which on activation of chymotrypsinogen loses the first 15 amino acids by selective proteolysis), which must be protonated to form a stabilizing salt bridge in the protein.
Note: A new theoretical computer program,
called
THEMATICS (theoretical microscopic titration curves) has been developed
to calculate the titration curves for all ionizable groups in a protein.
When performed on test proteins, those amino acids that showed anomalous
curves (flattened compared to normal titration curves) where usually found
in the active site of the protein. The flattened curves show that the
amino acid is partially protonated over a wider range of pH then
theoretically expected. The program can be used to predict active site
regions on protein of known structure but unknown function, and will be
useful in the emerging field of proteomics. (figure below from Proc. Natl.
Acad. Sci. USA, Vol. 98, Issue 22, 12473-12478, October 23, 2001)
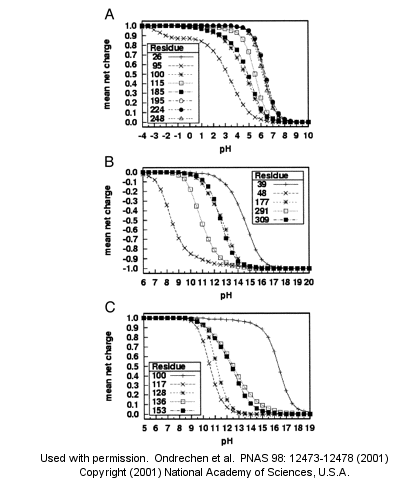
Fig. 1. Sample theoretical titration curves. Predicted mean net charge as a function of pH. (A) All of the histidine residues in the A chain of TIM: His-26 (+), His-95 (�), His-100 (*), His-115 (open square), His-185 (filled square), His-195 (open circle), His-224 (filled circle), and His-248 (). (B) Selected tyrosine residues of AR: Tyr-39 (+), Tyr-48 (�), Tyr-177 (*), Tyr-291 (open square), and Tyr-309 (filled square). (C) Selected lysine residues of PMI: Lys-100 (+), Lys-117 (�), Lys-128 (*), Lys-136 (open square), and Lys-153 (filled square). TIM, triosephosphate isomerase; AR, aldose reductase; PMI, phosphomannose isomerase. Remember that depending on the protein microenvironment, the pKa of a side chains like Asp can vary from 0.5 to 9.2!
- the enzyme - by chemical modification of specific amino acids, or through site-specific mutagenesis:
- modification of chyrmotrypsin (and many other proteases) with diisopropylphosphofluoridate (DIPF) modifies one of many Ser residues (Ser 195), suggesting that it is hypernucleophilic. and probably the amino acid which attacks the carbonyl C in the substrate, forming the acyl-intermediate.
Figure: diisopropylphosphofluoridate
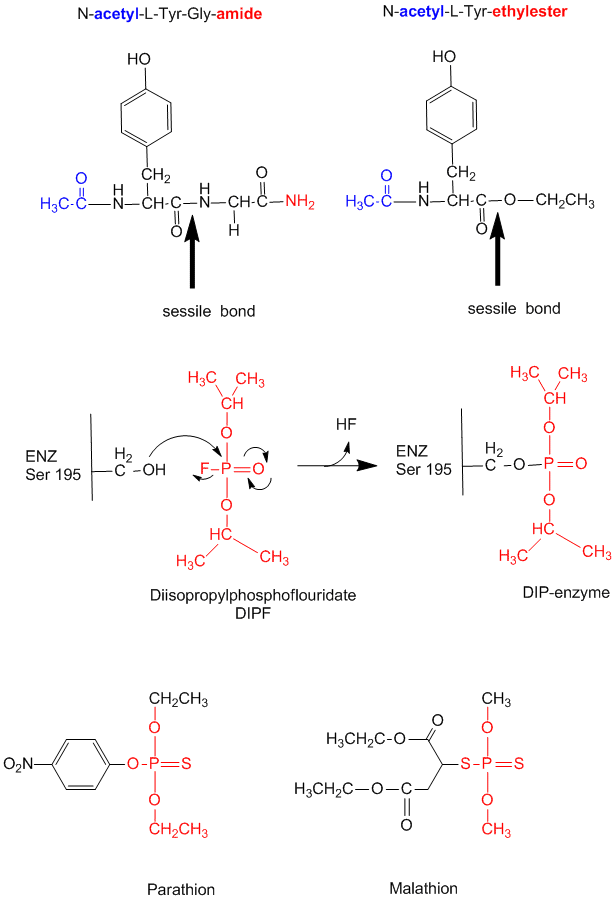
Malathion and ethyl parathion (organic phosphate pesticides) have similar structures and reactivities as DIPF, and selectively inhibit serine proteases in insects.
- modification of the enzyme with tos-L-Phe-chloromethyl ketone inactives the enzyme with a 1:1 stoichiometry which results in a modified His.
Figure: tos-L-Phe-chloromethyl ketone
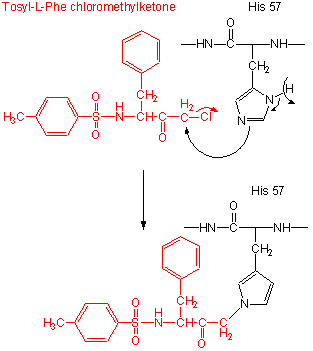
- comparison of the primary sequence of many proteases show that three residues are invariant: Ser 195, His 57, Asp 102.
- site-specific mutagenesis show that if Ser 195 is changed to Ala 195, the enzymatic activity is almost reduced to background.
Figure: Serine Protease Mechanism
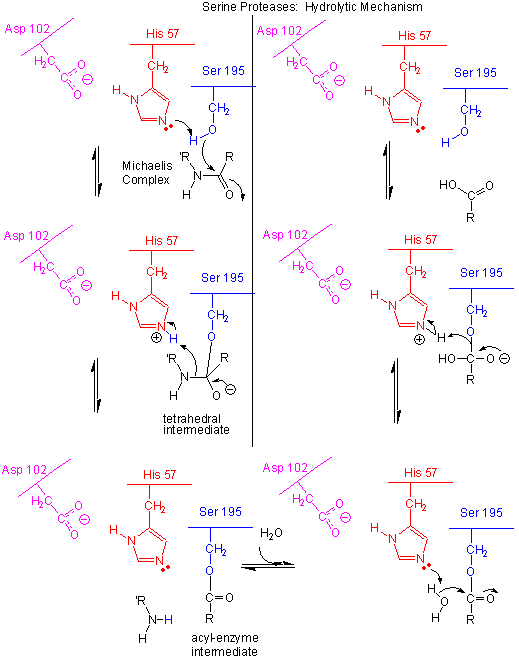
- The deprotonated His 57 acts as a general base to abstract a proton from Ser 195, enhancing its nucleophilicty as it attacks the electrophilic C of the amide or ester link, creating the oxyanion tetrahedral intermediate. Asp 102 acts electrostatically to stabilize the positive charge on the His.
- The oxyanion collapses back to form a double bond between the O and the original carbonyl C, with the amine product as the leaving group. The protonated His 57 acts as a general acid donating a proton to the amine leaving group, regenerating the unprotonated His 57.
- The mechanism repeats itself only now with water as the nucleophile, which attacks the acyl-enzyme intermediate, to form the tetrahedral intermediate.
- The intermediate collapses again, releasing the E-SerO- as the leaving group which gets reprotonated by His 57, regenerating both His 57 and Ser 195 in the normal protonation state. The enzyme is now ready for another catalytic round of activity.
- The mechanism for the first nucleophilic attack (by Ser) is the same as for the second (by water). The reverse mechanism of condensation of two peptide would be the reverse of the above mechanism, and is an example of the principle of microscopic reversibility.
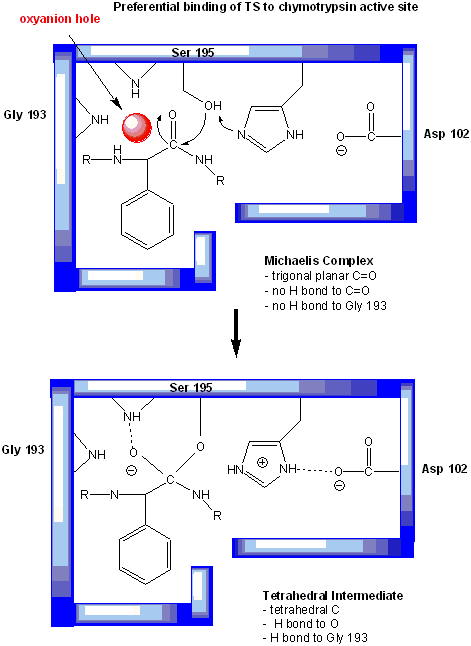
In short, all the catalytic mechanisms we encountered previously are at play in chymotrypsin catalysis. These include nucleophilic catalysis (with the Ser 195 forming a covalent intermediate with the substrates), general acid/base catalysis with His 57, and loosely, electrostatic catalysis with Asp 102 stabilizing not the transition state or intermediate, but the protonated form of His 57. An important point to note is that His, as a general acid and base catalyst, not only stabilizes developing charges in the transition state, but also provides a path for proton transfer, without which reactions would have difficulty in proceeding. One final mechanism is at work. The enzyme does indeed bind the transition state more tightly than the substrate. Crystal structures with poor "pseudo"-substrates that get trapped as partial tetrahedrally-distorted substrates of the enzyme and with inhibitors show that the oxyanion intermediate, and hence presumably the TS, can form H-bonds with the amide H (from the main chain) of Gly 193 and Ser 195. These can not be made to the trigonal, sp2 hybridized substrate. In the enzyme alone, the hole into which the oxyanion intermediate and TS would be placed is not occupied. This oxyanion hole is occupied in the tetrahedral intermediate.
Figure: oxyanion hole in serine proteases: TS stabilzation
Web Links:
![]() Jmol:
Updated Chymotrypsin:D-Leu-L-Phe-p-fluorobenzylamde complex
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Chymotrypsin:D-Leu-L-Phe-p-fluorobenzylamde complex
Jmol14 (Java) |
JSMol (HTML5)
![]() Jmol: Updated
Chymotrypin-Phenylethylboronate Inhibitor Complex
Jmol14 (Java) |
JSMol (HTML5)
Jmol: Updated
Chymotrypin-Phenylethylboronate Inhibitor Complex
Jmol14 (Java) |
JSMol (HTML5)
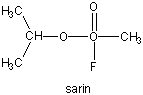
Many enzymes have active site serines which act as nucleophilic catalysts in nucleophilic substitution reactions (usually hydrolysis). One such enzyme is acetylcholine esterase which cleaves the neurotransmitter acetylcholine in the synapse of the neuromuscular junction. The transmitter leads to muscle contraction when it binds its receptor on the muscle cell surface. The transmitter must not reside too long in the synapse, otherwise muscle contraction will continue in an uncontrolled fashion. To prevent this, a hydrolytic enzyme, acetylcholine esterase, a serine esterase found in the synapse, cleaves the transmitter, at rates close to diffusion controlled. Diisopropylphosphofluoridate (DIPF) also inhibits this enzyme which effectively makes it a potent chemical warfare agent. An even more fluoride-based inhibitor of this enzyme, sarin, is the most potent lethal chemical agent of this class known. Only 1 mg is necessary to kill a human being.
Figure: sarin
B4. Proteases Mechanisms
Serine proteases are just one type of endoproteases. However, they are extremely abundant in both prokaryotes and eukaryotes. Protease A, a chymotrypin-like protease from Stremptomyces griseus, has a very different primary sequence than chymotrypsin, but its overall tertiary structure is quite similar to chymotrypsin, The positions of the catalytic triad amino acids in the primary sequences of the protein are very similar, indicating that the genes for the proteins diverged from a common precursor gene. In contrast, subtilisin, a serine protease from B. Subtilis, has both limited sequence and tertiary structure homology to chymotrypsin. However, when folded it also has a catalytic triad (Ser 221 - His 64 - Asp 32) similar to that of chymotrypsin (Ser 195 - His 57 - Asp 102).
![]() Jmol:
Updated Comparison of Chymotrypin and Subtilisin
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Comparison of Chymotrypin and Subtilisin
Jmol14 (Java) |
JSMol (HTML5)
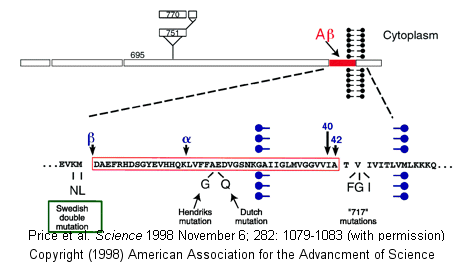
Proteases have multiple functions, other than in digestion, including degrading old or misfolded proteins and activating precursor proteins (such as clotting proteases and proteases involved in programmed cell death). In general, four different classes of proteases have been found, based on residues found in their active sites. Proteases can also be integral membrane proteins, and carry out their activities in the hydrophobic environment of the membrane. For example, aberrant cleavage of the amyloid precursor protein by the membrane protease presenillin can lead to the development of Alzheimers.
Figure: amyloid precursor protein
Classification of Proteases
| Class (active site) | Active Site Nucleophile | Location | Examples |
| Serine/Threonine Hydrolases | Ser/Thr | soluble | trypsin, chymotrypsin, subtilisin, elastase, clotting enzymes, proteasome |
| membrane | Rhomboid family | ||
| Aspartic Hydrolases | H2O activated by 2 Asps | soluble | pepsin, cathepsin, renin, HIV protease |
| membrane | β-secretase (BACE), presenilin I, signal peptide peptidase | ||
| Cysteinyl Hydrolases | Cys | soluble | bromelain, papain, cathespsins, caspases |
| membrane | ? | ||
| Metallo Hydrolases | H2O activated by 1 or 2 metal ions | soluble | thermolysin, angiotensin converting enzyme |
| membrane | S2P family |
| Glutamate Hydrolases | Glu | . | eqolysins (fungal) |
| Asparagine Lysases (EC4) (elimination rx which are self-cleavage and hence not catalytic) | Asn | . | Tsh autotransporter E. Coli |
β-secretase (BACE) is a membrane protein that contains two necessary Asp residues in its ectodomain (extracellular domain) which are used in the first cleavage of the N terminal domain of the beta amyloid precursor protein to release a soluble, N-terminal fragment of about 100,000 MW. γ-secretase, necessary for the second cleavage which frees the Aβ peptide is a heterotetramer composed of presenillin-1, nicastrin, APH-1 and PEN-2, and is located in neural plasma membranes and endoplasmic reticulum. The Aβ peptide moves to the extracellular side of the neural membrane where it aggregates. The remaining cytoplasmic part of the beta-amyloid precursor protein may regulate transcription. The presenillin subunit has protease activity. γ-secretase also cleaves another cell surface receptor protein, Notch. When this receptor has bound an extracelluar ligand, γ-secretase cleaves Notch within the cytoplasm, and the released fragment modifies gene transcription. The APH-1 subunit appears to inhibit presenilin protease activity while PEN-2 promotes it. Inhibiting γ-secretase would be an effect treatment for Alzheimers, but might have serious side effects since Notch processing would also be affected.
Figure: Cleavage of beta amyloid precursor protein: protease and cofactors
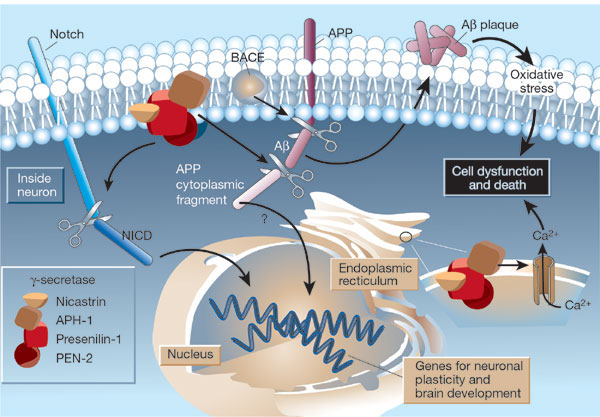
|
The γ-secretase protein quartet, and its roles in brain development and Alzheimer's disease. Presenilin-1, nicastrin, APH-1 and PEN-2 form a functional γ-secretase complex, located in the plasma membrane and endoplasmic reticulum (ER) of neurons. The complex cleaves Notch (left) to generate a fragment (NICD) that moves to the nucleus and regulates the expression of genes involved in brain development and adult neuronal plasticity. The complex also helps in generating the amyloid β-peptide (Aβ; centre). This involves an initial cleavage of the amyloid precursor protein (APP) by an enzyme called BACE (or β-secretase). The γ-secretase then liberates Aβ, as well as an APP cytoplasmic fragment, which may move to the nucleus and regulate gene expression. Mutations in presenilin-1 that cause early-onset Alzheimer's disease enhance γ-secretase activity and Aβ production, and also perturb the ER calcium balance. Consequent neuronal degeneration may result from membrane-associated oxidative stress, induced by aggregating forms of Aβ (which create Aβ plaques), and by the perturbed calcium balance. Reprinted by permission from Nature: Mattson, M. Nature. 422, 385-387 (2003) |
How do integral membrane protease catalyze the hydrolysis (using water) of transmembrane domains in proteins, given the hydrophobic environment of the bilayer? The rhomboid class of membrane proteases, which are found in prokaryotic and eukaryotic cells, is one of the most conserved membrane proteins in nature. Sequence comparison of rhomboid proteins shows a possible catalytic serine residue that is buried to the same depth in the bilayer as the cleavage site for rhomboid protein substrates. In addition the proteases share conserved amino acids similar to the catalytic triad of soluble serine proteases, with the critical amino acid localized on different membrane-spanning regions of the protein. The membrane triad amino acids are Ser, His and Asn (instead of Asp as in classical serine proteases). Classical serine protease inhibitors prevent cleavage of membrane protein substrates, but given the difficulty of reconstituting a functional purified rhomboid protein in vitro, the identity of specific amino acids in the catalytic mechanism is inferential at present.
Helical wheel diagrams of putative transmembrane domains of the rhomboids (determined through hydropathy plots) show several to be amphiphilic. This might allow water, necessary for hydrolysis, to enter into the catalytic active site of the enzyme, although as was noted before, water has a reasonably high permeability through a membrane bilayer. The chief requirement for protein substrates of rhomboids is the presence of a transmembrane domain in the target protein. No specific amino acid sequence seems to be required for specificity of one particular substrate, the drosophila transmembrane protein spitz found in Golgi membranes. On cleavage of this protein, the remaining part of the protein is released as a water soluble protein to the lumen of the Golgi where it can eventually be released from the cell. The soluble protein fragment that is released from the cell contains an epidermal growth factor domain.
Recently the structure of a rhomboid protease, GlpG, from E. Coli, was determined. This transmembrane protein has 6 transmembrane helices. The enzyme has a polar active site at the bottom of a V-shape opening situated laterally in the membrane. Active site His and Asn residue and many water molecules are deep in this V-shaped cleft well below the surface of the membrane. Access to the transmembrane strand of the protein substrate is blocked by a loop, which must be gated open to allow substrate access between the V-shaped gap between helices S1 and S3. Ser 201 (nucleophile) and His 254 (general base/acid) are essential for activity.
![]() Jmol:
Updated E. Coli GlpG rhomboid intramembrane protease
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated E. Coli GlpG rhomboid intramembrane protease
Jmol14 (Java) |
JSMol (HTML5)
B5. Protease Activation and the Proteasome
Protease are potentially dangerous if their activity is not regulated. A common method to avoid unwanted protease activity is to activate the enzyme from an inactive precursor called a zymogen. The precursor is often called a proenyzme. Limited but regulated proteolysis of the proenzyme by either a different protease or by autoproteolysis leads to activation of the proteolytic activity of the zymogen. One important example is activation of procaspases to active caspases, which are calcium activated cysteine proteases that are homodimers. Activation of caspases initiates programmed cell death. Cancer cells which are immortal have found ways to inhibit procaspase activation. Hence a possible cancer therapy could involve drug-induced activation of procaspases. Wolan et used high-thoroughput screening to identify compounds that promote the activation of procaspase-3 at physiological conditions and concentrations. A dozen compounds were found to promote such activity, and their ability to activate other similar enzymes in the procaspase family were explored. Active caspases appear to be in equilibrium between an active state and an inactive one more similar to the inactive zymogen. Small drug that bind preferentially to the active state would shift the equilibrium from the inactive state to the active state. Likewise it might bind to the inactive zymogen and promote an "active" conformation of the zymogen leading to the actual activation of the zymogene by autoproteolysis. Wolan et al discovered that procaspase was able to undergo a conformational change with the addition of a specific small molecule activator (referred to as 1541) which made the “on (active)” conformation more likely, and therefore encouraged self-activation.
Proteases are found in both extracellular (digestive tract, blood, extracellular matrix), membrane, and and intracellular locations. As mentioned previously, one role of intracellular proteases is to degrade "older" and chemically damaged proteins. One of the main proteases involved in such intracellular proteolysis is the large protein complex called the proteasome. It consist of three large structures
-
one 700,000 MW 20S complex which contains 14 different subunits arranged in 4 rings of 7 subunits (2 copies of each subunit). The rings are stacked on each other. The outer two rings contain α subunits while the middle two contain β subunits. ATP-dependent cleavage of protein substrates occurs within the complex, with N terminal Ser or Thr OH groups acting as nucleophiles.
-
two 19S complex which cap both ends of the 20S complex (not unlike the structure of the E. Coli chaperone complex GroEL/ES.
Proteins destined for cleavage by the proteasome must first be chemically modified through attachment of multiple copies of the 8,500 MW protein ubiquitin, a highly conserved protein found ubiquitously in eukaryotes. (We modeled this protein in the first lab using VMD and NAMD.) The carboxyl group of the C-terminal Gly residue of ubiquitin forms an amide link to the side chain amine group of Lys residues in the protein targeted for degradation. The resulting link is an isopeptide bond since the N terminal of the target protein is not used in the amide bond. Three different proteins are involved in the ubiqutinylation of the target protein, including E1 (ubiquitin-activating enzyme which requires ATP), E2 (ubiquitin conjugating enzyme) and E3 (ubiquitin-protein lyase). Once attached, a side chain Lys of ubiquitin can form another isopeptide bond to a C-terminal carboxyl group of another ubiquitin, forming a growing ubiquitin chain on the target protein. Proteins with 4 or more linked ubiquitins are better substrates for the proteasome. Proteins with short half-lives (those with certain amino terminal amino acids like arginine or leucine, or enriched in Pro (P), Glu (E), Ser (S), and Thr (T) - (PEST) appear to be better targets for the ubiquitin pathway and subsequent degradation by the proteasome.
Proteasome activity is intimately related to health and disease. A major role of the proteasome occurs in immune recognition of self and nonself. The immune system must be able to recognize a virally infected or tumor cell (both self cells expressing foreign or aberrant proteins) as well as foreign cells like bacteria, which can be engulfed by immune cells such as macrophages. Proteasome involvement occurs when viral, tumor, or bacterial proteins are degraded to short peptides, which bind intercellular major histocompatability proteins (MHC) proteins and are translocated to the cell membrane. Peptide/MHC complexes are displayed on the cell surface and are recognized by receptors on immune cells (specifically T cells). Self cells are not recognized by T cells since the peptides in the peptide:MHC complex are self peptides derived from normal proteins. The T cell receptor recognizes determinants on both the MHC protein and the presented peptide.
![]() animation
of protein processing and display of peptides by MHC proteins on cell
surface
animation
of protein processing and display of peptides by MHC proteins on cell
surface
![]() HHMI
animation of the ubiquitin and the proteasome
HHMI
animation of the ubiquitin and the proteasome
Nonrecognition of self peptide:MHC complexes prevents the immune system from targeting normal healthy cells. Autoimmune diseases arise when the T cell receptor recognizes presented self peptides.
The ubiquitin/proteasome pathways have been linked to disease manifestation in many neurodegenerative diseases like Alzheimers, Huntington's disease (which involves the aberrant folding of the Huntington protein which contains an expanded poly-Glu domain), and Parkinsons. The degradation pathway is involved in many other normal cellular functions including gene transcription and programmed cell death.
![]() Jmol:
Updated Mammalian Proteasome - (1IRU)
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Mammalian Proteasome - (1IRU)
Jmol14 (Java) |
JSMol (HTML5)
B6. Motion and Enzyme Catalysis
Benkovic and Hammes-Schiffer have offered an in-depth review of methods used by enzymes in catalysis. They divided their analysis into a biochemical and theoretical frameworks. From a biochemical viewpoint, enzyme catalysis was thought to result from binding of substrate followed by activation of the substrate by the enzyme. The substrate might bind in a particularly reactive conformation in a lock-and-key fashion, or be distorted by the enzyme on binding, making it more susceptible to bond making/breaking. In an energy diagram for the latter case, the bound distorted substrate has higher energy than the free substrate, and is an example of substrate (ground state) destabilization. With the theoretical work of Pauling and development of catalytic antibodies by Lerner, the idea than enzyme functions by binding the transition state with higher affinity than the substrate became accepted. Evidence for tighter transition state binding was found for serine proteases (through stabilization of the tetrahedral oxyaninon intermediate) and lysozyme (through stabilization of the developing oxocarbenium ion (positive charge) by adjacent Asp and Glu residues (both of which are described above). Quantitative increases in catalytic hydrolysis of phosphoesters and diesters of 1015 to 1017 by some phosphatases and nucleases over the uncatalyzed, spontaneous hydrolysis in aqueous solutions are known. Assuming that the ratio of the catalyzed rate at low S (kcat/Km with units of M-1s-1) versus the noncatalyzed rate (pseudo first order with using of s-1) gives a measure of the effective concentration of substrate in the noncatalyzed rate that would give the same rate as the catalyzed rate, effective concentrations reach as high as 1024 for enzymes like orotic acid decarboxylase. What is the source of this catalytic power? Much evidence has been accrued to partition catalysis into contributions from general acid/base catalysis, nucleophilic and electrostatic catalysis, as well as binding effects that enhance catalysis. Solvent effects also show that for some reactions, solvation slows the reaction, implying that desolvation of the substrate during substrate-enzyme binding affects the reaction rate. Remember that the active site in the E-S complex is devoid of most water molecules that are present in the free enzyme. Finally the enzyme may have the correct 3D framework that positions all side chains involved in binding in catalysis in optimal positions, which may have the effect of selecting only catalytically-competent conformations of the substrate (i.e. those the most resemble the transition state). These contributions suggest that movement of critical residues in the active site is required, which positions the substrate and catalytic residues in optimal positions for catalysis. The kinds of motions (which can be modeled in molecular dynamics simulations) are described in the table below.
Table: Time Scales of Dynamic Events in Enzyme Catalysis
| Motion | Approx. Time Scale - log(s) |
| bond vibration | -14 to -13 |
| proton transfer | -12 |
| hydrogen bonding | -12 to -11 |
| elastic vibration of globular region | -12 to -11 |
| sugar repuckering | -12 to -9 |
| rotation of side chains at surface | -11 to -10 |
| torsional libration of buried group | -11 to -9 |
| hinge bending at domain interfaces | -11 to -7 |
| water structure reorganization | -8 |
| helix breakdown/formation | -8 to -7 |
| allosteric transitions | -5 to 0 |
| local denaturation | -5 to 1 |
| rotation of medium-sized interior sidechains | -4 to 0 |
from Benkovic, S and Hammes-Schiffer, S. Science. 301, pg 1197 (2003)
Warshel argues that electrostatic stabilization of the transition state is the predominate mechanism for tighter binding of the transition state than the substrate to the enzyme. Hydrophobic interactions would facilitate proper orientation of the substrate/transition state in the active site pocket, which is preorganized in effect by the structure of the enzyme. Other have argued that quantum mechanical tunneling (of hydrides, protons, or electrons) is a main mechanism to surmount energetic barriers in enzyme-catalyzed reactions. Using theoretical and molecular dynamic simulations, Gao et al argue that tunneling, a way to overcome classical energy barriers, plays a much smaller role that reducing the barrier height (activation energy). Warshel notes that tunneling effects can also occur in solution in the absence of enzyme, making it all the more important to compare reactions mechanisms in the presence and absence of enzyme. Yet proteins motions that move catalytically important side chains toward the substrate/transition state may allow both preferential electrostatic stabilization of the transition state and quantum mechanical tunneling. Yet another idea states that enzymes must create new bonds to substrate/transition state that could not be available in the absence of the enzyme. One such type of bond is a low-barrier hydrogen bond, in which a hydrogen on an electronegative atom is involved in strong H bonds to two different hydrogen bond acceptors simultaneously. Finally, substrate may adopt near-attack conformations (NAC), which more approximate the bound transition state, and it is the binding of the NAC to the enzyme, not lowering the activation energy, that contributes to the enzyme catalyzed rate enhancement.
B7. Quantum Mechanisms
TBA
B8. Links and References
![]() The
Enzyme Function Initiative is
developing such tools to predict in vitro enzymatic and in vivo
physiological functions of unknown enzymes.
The
Enzyme Function Initiative is
developing such tools to predict in vitro enzymatic and in vivo
physiological functions of unknown enzymes.
![]() Uniprot,
a web base resources that hold protein sequence and functional
information, has over 44 million protein sequences (derived from nucleotide
sequences) and most have no well defined protein function.
Uniprot,
a web base resources that hold protein sequence and functional
information, has over 44 million protein sequences (derived from nucleotide
sequences) and most have no well defined protein function.
![]() Metabolic
Docker: uses molecular docking as a basis for predicting the
function of enzymes. It supports docking both ground state and high energy
intermediate forms of metabolites and commercially available compounds to
protein structures
Metabolic
Docker: uses molecular docking as a basis for predicting the
function of enzymes. It supports docking both ground state and high energy
intermediate forms of metabolites and commercially available compounds to
protein structures
Other Types of Enzymes
The three enzymes studied above are all hydrolases - enzymes that catalyze the hydrolysis of bonds (either amide or acetal). This is only one class of six different reaction types that have been categorized by the Enzyme Commission of the International Union of Biochemistry and Molecular Biology. The six types (all external links) include:
EC1: Oxidioreductases - oxdiation/reduction reactions (we will discuss these in Chapter 8B)
EC2: Transferases - acyl, glycosyl, 1C, N, O, aldehydes, ketones, etc
EC4: Lyases - cleavage of C-C, C-O, C-N, C-S, etc. bonds
EC5: Isomerases - racemases, epimerases, cis-trans isomerases
EC6: Ligases - form C-C, C-O, C-N, etc bonds
Other Links
![]() Enzyme
Nomenclature Database: Interactive site to search information on
enzymes using EC system of nomenclature.
Enzyme
Nomenclature Database: Interactive site to search information on
enzymes using EC system of nomenclature.
![]() BRENDA:
(Brauschweig Enzyme Database) Comprehensive Enzyme Information System
BRENDA:
(Brauschweig Enzyme Database) Comprehensive Enzyme Information System
![]() KEGG
PATHWAY: collection of manually drawn pathway maps
representing our knowledge on the molecular interaction and reaction
networks using KEGG, Kyoto
Encyclopedia of Genes and Genomes
KEGG
PATHWAY: collection of manually drawn pathway maps
representing our knowledge on the molecular interaction and reaction
networks using KEGG, Kyoto
Encyclopedia of Genes and Genomes
![]() FMM
(From Metabolite to Metabolite) - reconstructs metabolic pathways from one
metabolite to another
FMM
(From Metabolite to Metabolite) - reconstructs metabolic pathways from one
metabolite to another
References
- Wolan, D. et al. Small-Molecule Activators of a Proenzyme. Science 326, 853 (2009)
- Wang, Y. et al. Crystal structure of a rhomboid family intramembrane protease. Nature. 444, 179 (2006)
- Freeman, M. Proteolysis within the membrane: rhomboids revealed. Nature Reviews: Molecular Cell Biology. 5, p 188 (2004)
- Borman, S. Much ado about enzyme mechanisms. C&EN. pg 35 (Feb 23, 2004)
- Garcia-Cioloca, M. Goa, J., Karplus, M. and Truhlar, D. How Enzymes Work: Analysis by modern rate theory and computer simulation Science. 303, pg 186 (2004)
- Benkovic, St. & Hammes-Schiffer, S. A Perspective on Enzyme Catalysis. Science. 301, pg 1196 (2003)
- Takasugi, N. et al. The role of presenilin cofactors in the γ-secretase complex. Nature. 422, pg 438 (2003)
- Weihofen et al. Identification of Signal Peptide Peptidase, a Presenilin-Type Aspartic Protease. Science, 296, pp. 2156, 2215,
- Vocadlo. D. et al. Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature. 412. pg 835 (2001)
- Walsh, C. Enabling the Chemistry of Life. Great review article on enzymes mechanisms. Nature. 409, pg 226 (2001)
- Koeller and Wong. Enzymes for Chemical Synthesis. Nature 409. pg 232 (2001)
- Simeonov et al. Blue-Fluorescent Antibodies. Science. 290, pg 286, 307 (2000)
- Huntington et al. Structure of a serpin-protease complex shows inhibition by deformation. Nature. 407, pg 923 (2000)
- New Way to Study Closely related proteins (remodeling proteins to make them more susceptible to inhibition) Science 289. pg 2029 (2000)
- Vocadlo et al. Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature. 412. pg 835 (2001)
- Istan and Deisenhofer. Structural Mechanism for Statin Inhibition of HMG-CoA Reductase. Science. 292, pg 1160 (2001)
- Heine et al. Observations of Covalent Intermediates in an Enzyme Mechanism at Atomic Resolution. Science 294. pg 369 (2001)
- Carpenter et al. Structure of dehydorquinate synthase reveals an active site capable of multi-step catalysis. Nature. 394, pg 299 (1998)
- Kohen et al. Tunnel Vision (on why activity of therophilic enzymes (>60oC) is low or absent at mesophilic temperatures (< 40oC) - from reduction of flexibility of thermophilic enzymes at mesophilic temperatures - quantum tunneling explanation). Nature. 399, pg 417, 496 (1999)
- Finnin et al. Structure of a histone deacetylase homologue bound to the TSA and SAHA inhibitors (and mechanism). Nature. pg 189, September 1999.
- Ondrechen. THEMATICS: A simple
computational predictor of enzyme function from structure. Proc. Natl.
Acad. Sci. USA, 98, pg 12473 (2001 )
 Navigation
Navigation
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 7B: Mechanisms of Enyzme Catalysis

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.